ACE-GCN
Adsorbate Chemical Environment-based Graph Convolutional Network
There are plethora of reactions wherein the material (catalyst) aiding the reaction undergoes phhysical and/or chemical transformation under the reaction conditions (in-operando). Heterogeneous catalytic reactions are sensitive to atomic-scale complexities arising under in-operando conditions, such as variations in surface morphology, composition, and adsorbate-adsorbate interactions. More so, presence of irregular geometries and complex material arrangement, present for the catalyst, expands the possible solution space of possible catalyst models for the reaction. As a result, the difficulty in systematically generating and analyzing the surface representations, make atomic-model development non-trivial. To address this challenge, a graph network-based enumeration and prediction strategy is explored.
Adsorbate Chemical-Environment-based Graph Convolution Neural Network (ACE-GCN), is a versatile framework with the ability to encode atomic configurations comprising of diverse adsorbates, binding locations, coordination environments, and variations in the substrate morphology. This workflow is used to generate and rank surface adsorbate configurations for reactions which are shown to be affected by the presence of high adsorbate surface coverage. Through this study we inch closer towards realizing the deam of better describing the molecular-level functioning for complex materials and process.
Background
Encoding catalyst surfaces for machine learning-based screening is a fairly recent expansion of the machine learning (ML) methods in computational material modeling. Exciting advances in database architecture, feature abstractions, and compute power have now made it possible to consider avenues of encoding complex information. Thanks to these advances, researchers are now considering catalysts models that are much wider in composition space, and modeling complex reaction networks that were hitherto deemed challenging. In most of these model development endevors the challenges are: (1) Feature representation of the atomic models which are machine readable and (2) Systematically generating the training dataset with minimal bias from the user to ensure exhaustive sampling of the search space.
One way of addressing this challenge is to use graph-networks for presenting the atoms and their bonding environment. This idea has been implemented in understanding social interactions, relationship between entities as seen in Indian spice usage, and in modeling, predicting molecular properties of small molecules which intuitively are condusive to such graphical network representation.
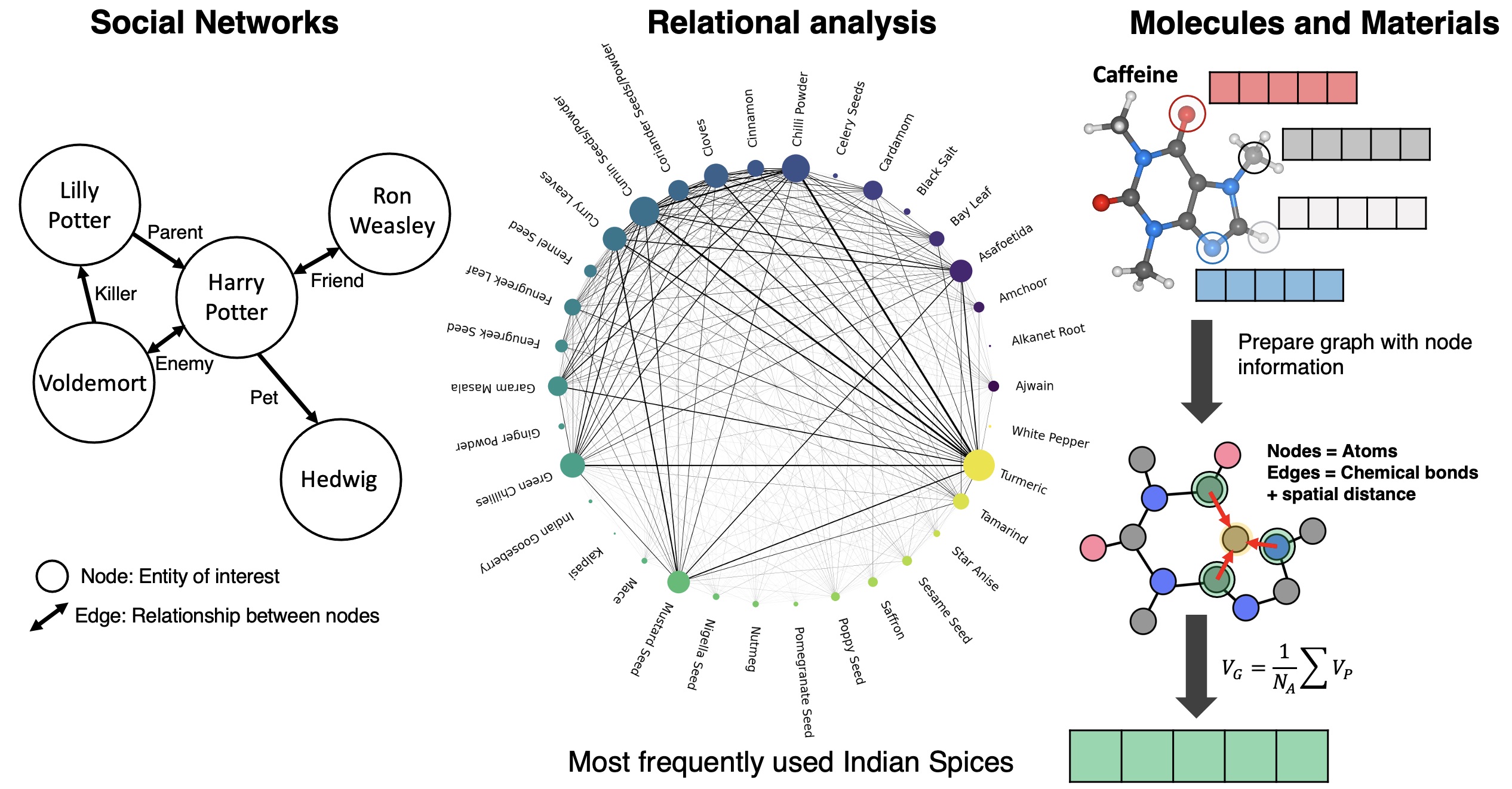
In this work we build upon the idea of graph network representation and use sub-graphs to represent high coverage coverage representations. Every sub-graph is generated using the chemical environment of each unique adsorbate in the system.
ACE-GCN
Adsorbate Chemical Environment-based Graph Convolutional Neural Network (ACE-GCN, for short) is a framework to help systematically generate, encode, and predict energetics of the surface models to help navigate the phase space to find stable surface representations.
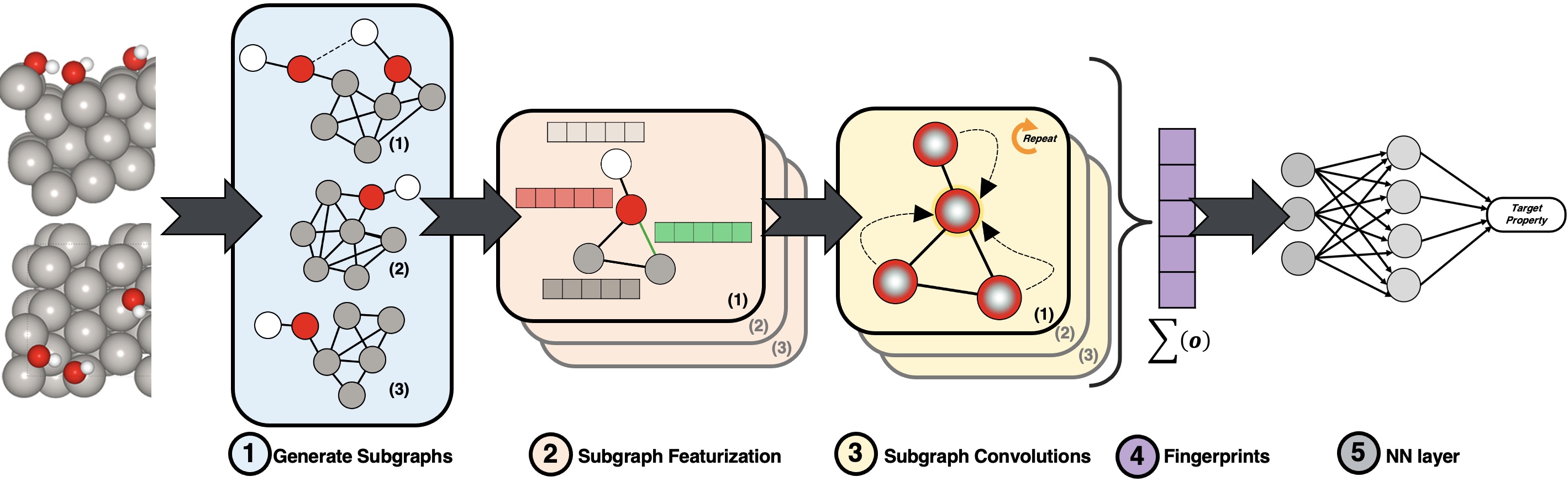
ACE-GCN leverages the expressive power of graph networks to encode and learn the mapping of electronic and geometrics features of the catalyst surface to its thermodynamic stability. Once trained, the model can be used to rank new surface representations and quickly select stable candidates.
The atomic-scale catalyst models and computation tools proposed through this work can serve as a starting point for developing a detailed description of complex catalyst surfaces under in-operando conditions, ultimately leading to fundamental insights into the factors that govern the functioning of heterogeneous catalysis in chemically complex environments.
Application to heterogeneous catalysis
We utilize this workflow to successfully sample the large number of atomic configurations to identify the most relevant surface representation stable reaction conditions. We demonstrate the utility of the workflow to determine the relevant high coverage configurations for the cases of adsorbed NO* on Pt3Sn (111) surface and that of adsorbed OH* on irregular Pt (221).
The first case presents complexities because of the presence of an alloy and the strong binding nature of NO, while the second case presents challenges arising from the presence of step defects and the ability of OH to form inter-molecular hydrogen bonds. Using these two test cases as representative examples for likely challenges when developing computational models, we demonstrate the generalizability of our workflow to identify the most relevant atomistic configurations by utilizing only 10% of the total possible configurations.
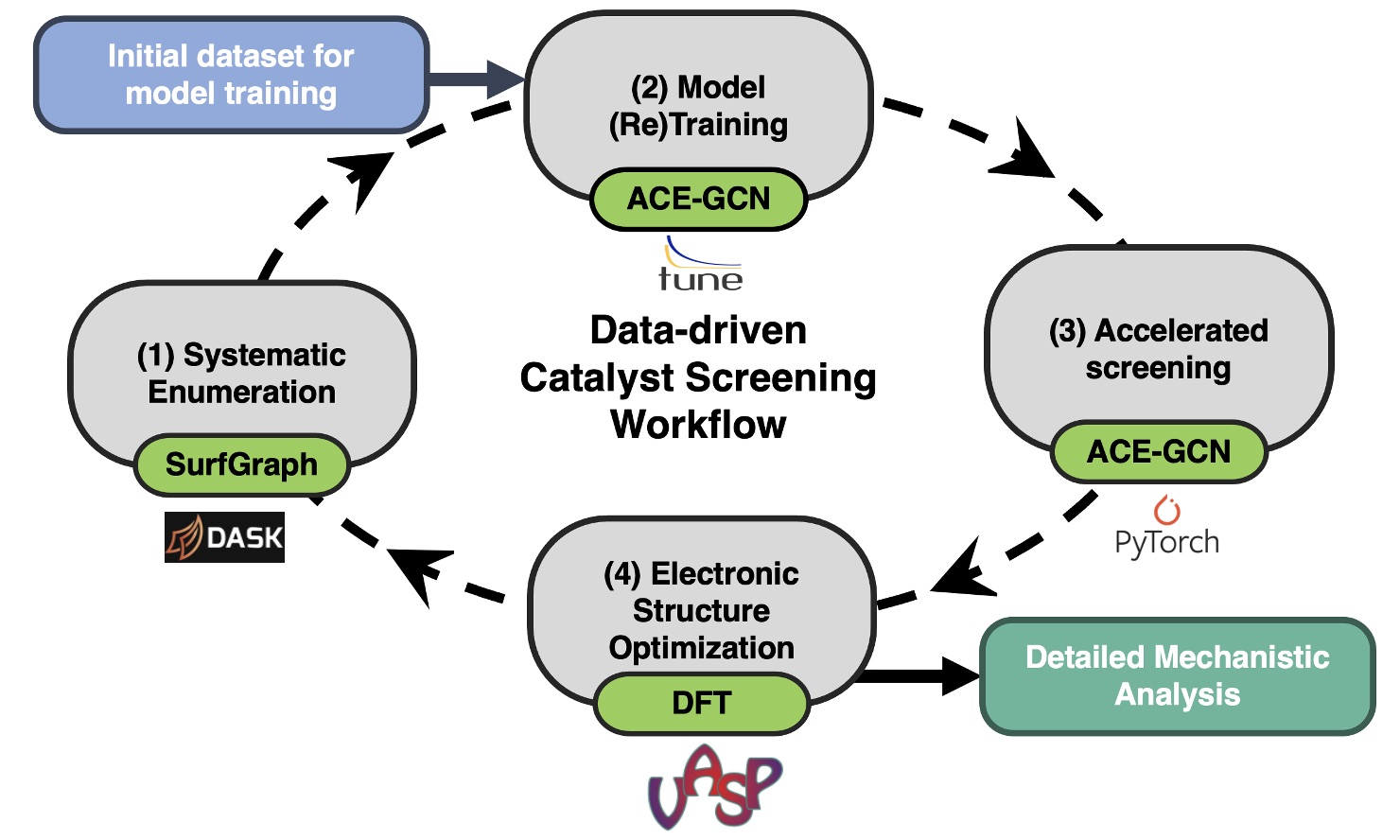
This approach can serve as a starting point for developing detailed atomic description of complex catalyst surfaces under in-operando conditions, ultimately leading to fundamental insights into factors that govern heterogeneous catalysis in structurally and chemically complex environments.
Future Reading:
Xie, T. & Grossman, J. C. Crystal Graph Convolutional Neural Networks for an Accurate and Interpretable Prediction of Material Properties. Phys. Rev. Lett. 120, 145301 (2018). Blog
Goodall, R.E.A., Lee, A.A. Predicting materials properties without crystal structure: deep representation learning from stoichiometry. Nat Commun 11, 6280 (2020).
Back, S. et al. Convolutional Neural Network of Atomic Surface Structures to Predict Binding Energies for High-Throughput Screening of Catalysts. J. Phys. Chem. Lett. 10, 4401–4408 (2019)
Lym, J., Gu, G. H., Jung, Y. & Vlachos, D. G. Lattice convolutional neural network modeling of adsorbate coverage effects. J. Phys. Chem. C 123, 18951–18959 (2019).
Schmidt, D. J.; Chen, W.; Wolverton, C.; Schneider, W. F. Performance of Cluster Expansions of Coverage-Dependent Adsorption of Atomic Oxygen on Pt(111). J. Chem. Theory Comput. 2012, 8, 264−273
Chowdhury, A. J. et al. Prediction of Adsorption Energies for Chemical Species on Metal Catalyst Surfaces Using Machine Learning. J. Phys. Chem. C 122, 28142–28150 (2018).